The New York Academy of Sciences and the Shanghai Association for Science and Technology are teaming up to advance science and innovation on a global scale.
Published March 12, 2024
By Nick Fetty
Nicholas B. Dirks, President and CEO of The New York Academy of Sciences, shakes hands with Zhiqiang Han, Executive Vice President of the Shanghai Association for Science and Technology, after signing a memorandum of understanding for a new collaboration between the two institutions.
Science diplomacy took a significant step forward recently, when Nicholas B. Dirks, President and CEO, The New York Academy of Sciences met with representatives from the Shanghai Association for Science and Technology (SAST) in Shanghai, to discuss hosting international science and technology forums. The resulting agreement codified a partnership dedicated to advancing long-term collaborations between the two institutions, as well as collaboration on youth scientific and technological education and innovations.
“Global issues require global solutions,” said Dirks. “This new partnership will be an excellent way for us to strengthen the relationship with our partners in Shanghai and other parts of China where promising STEM education, research and innovation is taking place. Given the current geopolitical climate, it is critical for us to develop these kinds of international collaborations to advance solutions for the public good globally.”
This sentiment was echoed by Professor Zhang Jie, President of the SAST. “A small step today is a giant leap for the future to promote scientific and technological cooperation between China and the United States, and even globally,” said Zhang. “Through the collaboration, we hope that more American scientists and even global scientists will understand Shanghai and China better, come to Shanghai and China, and carry out borderless scientific and technological cooperation.”
Enhancing the Junior Academy
A follow-up visit by Meghan Groome, PhD, Senior Vice President of Education at the Academy, advanced the conversation on potential in-person and virtual collaborations for students studying STEM (science, technology, engineering and mathematics). The Academy’s award-winning Junior Academy provides a scalable and impactful way for students to participate in virtual exchange programs. The Academy plans to expand its partnership with students of Shanghai through increased outreach to schools in the region.
Nicholas B. Dirks, President and CEO of The New York Academy of Sciences, and Jie Zhang, President of the Shanghai Association for Science and Technology.
While in Shanghai, Dr. Groome also addressed the Deeper Learning China conference, an event dedicated to building a community of educators in China focused on Project Based Learning. in collaboration with Deeper Learning Global. Dr. Groome encouraged educators to take a light-hearted approach to implementing AI in the classroom, while building their knowledge base about the strengths and weaknesses of different AI products. She also discussed future plans with the Deeper Learning China leadership, to explore ways to expand The Junior Academy, a “game changing” student collaboration and research network, to more schools in China.
The Role of Artificial Intelligence
Groome also participated in the first “China-United States Seminar focused on the Vision and Future Feasibility of Artificial Intelligence in Arts Education”. At the Nine Trees Performing Arts Complex in Shanghai, she spoke about the importance of understanding the impact AI will have on our societies as a whole, including the arts.
“Although my focus is STEM, I’m increasingly relying on my background in the arts to help explore and explain these large, technological shifts in our lives. We can no longer teach in siloes but must expand how the integration, or convergence of subjects can lead to a better understanding of our changing world,” she told the audience. “I believe that AI can expand access to the highest quality arts [and STEM] education through AI-driven software and instruments. Like with sports, AI can enhance our practice time, providing us with expert feedback anytime, anywhere. Finally, it can dramatically enhance our creativity and allow us to collaborate like never before.”
Vice Chairman and Chief Scientific Officer PepsiCo
This challenge fostered a multitude of innovative ideas, and we could not be happier to support this endeavor. By addressing critical undernutrition at home and with women, the Solvers have played a vital role in the health of mothers, their children, and future generations.
Vice President of Technology Strategy and Innovation Lockheed Martin
We’re proud to work with the New York Academy of Sciences on this initiative and believe the ideas brought forward could positively influence how Lockheed Martin integrates advanced technologies for aerospace and defense capabilities.
Eugene Higgins Professor of Electrical Engineering, Professor of Applied Physics Columbia University Blavatnik National Awards Scientific Advisory Council
There are a few awards for young scientists, but almost all of them are based on proposals that you submit, and not on the actual work that you do as a young scientist. The Blavatnik Awards is true recognition of the work of young scientists; it is unique in that sense. There is no equivalent.
As Chair of the Board of Governors, Jill and I are thrilled to support The New York Academy of Sciences’ renewed drive to create a positive impact and encourage science-informed decision-making to address the most challenging problems of our times.
Chief Medical Officer and Head of Worldwide Medical & Safety Pfizer Board of Governors The New York Academy of Sciences Executive Board member International Science Reserve
Pfizer has been a proud partner of the Academy for nearly 20 years, supporting multiple shared areas of interest, from supporting science and scientists through every stage of their careers, to driving a preparedness framework to empower scientists to act when the next global crisis hits.
Can we stop the pain? It may be the oldest question in medicine, and it remains one of the most important. But with chronic pain afflicting billions of people worldwide, and few effective treatments besides highly addictive opioids, researchers are still searching for better answers.
On May 3-4, the New York Academy of Sciences, in collaboration with Science Translational Medicine, convened the Advances in Pain conference. Across the meeting’s two keynote presentations, nine sessions of talks, and concluding panel discussion, leading experts in many branches of pain research discussed the field’s biggest challenges and latest developments.
Highlights
Specific ion channels on neurons, such as Nav1.7, are critical components of pain sensing and potential drug targets for new analgesics.
Several novel laboratory models are revealing new details of nociception, or pain sensing.
Large databases of genetic and clinical records are helping researchers link specific genes with common pain conditions.
Neuroimaging and sleep studies may offer objective ways to measure the severity of chronic pain.
New mechanistic data are pointing researchers toward novel strategies for analgesic drug development.
A subset of gut epithelial cells is critical for sensing visceral pain.
The immune system links tightly to pain sensation, through multiple mechanisms scientists are now beginning to uncover.
Data mining reveals subsets of neurons with distinct responses to nerve injury, including chronic pain.
Understanding sex and ethnic differences in pain perception requires new strategies in experimental design and data analysis.
Besides neurons, Schwann cells can also carry pain signals.
Novel drug discovery platforms and trial designs can accelerate the development of new analgesics.
Part 1
Speakers
David Bennett, MB, PhD Oxford University, Nuffield Department of Clinical Neurosciences
Sarah E. Ross, PhD University of Pittsburgh
Jing Wang, MD, PhD NYU Langone Health
Tuning into the pain channel
A life free of pain may sound ideal, but as David Bennett explained in the meeting’s opening keynote presentation, individuals with defects in pain sensing often suffer tremendous difficulties. Describing one 26-year-old man with such a condition, Bennett explained that “he had pretty much fractured every long bone in his body, he is stunted because he’s destroyed all the growth plates … and had severe burns and mouth injuries.” The patient’s sister, who had the same condition, died of undiagnosed sepsis.
Genetic analysis revealed that the patient had a rare set of loss-of-function mutations in the gene for Nav1.7, a sodium ion channel expressed in nociceptors, or pain sensing neurons. Using a sophisticated cell culture system that mimics pain signaling through nociceptors, Bennett and his colleagues have characterized Nav1.7 in detail, and determined that it acts early in the pain signaling process, amplifying the electrical signal in the nociceptors to ensure that it’s relayed to the central nervous system.
Patients with gain-of-function mutations that make Nav1.7 overactive have the opposite problem: incurable chronic pain. Bennett’s team studied the Nav1.7 mutations in these patients, and discovered that the degree of the biochemical defect in a patient’s channel proteins correlates directly with the time of onset of their pain condition.
Based on his findings in patients with these rare, extreme pain disorders, Bennett hypothesized that Nav1.7 could also drive more common conditions. As rates of diabetes skyrocket globally, millions of people are developing diabetic neuropathy, which causes chronic pain only in a subset of patients. In an effort to determine what distinguishes painful from pain-free diabetic neuropathy, Bennett’s team looked at Nav1.7 gene sequences for patients with the condition.
“The rare variants in Nav1.7 seemed to cluster much more in the painful versus the painless diabetic neuropathy groups, so this is now acting as a risk factor, in the sense that these people did not experience [chronic] pain prior to developing diabetes,” Bennett says.
Some variants of Nav1.7 apparently predispose people to develop chronic pain, but the condition doesn’t manifest itself until a second event, such as diabetes, triggers it. A closer look at clinical testing results in these patients revealed that those with the rare variants were also more sensitive to certain stimuli, such as burning pain and pressure pain.
Nav1.7 isn’t the only ion channel involved in pain, though. The researchers have also identified strong associations between pain disorders and mutations in the related channel proteins Nav1.8 and Nav1.9, highlighting the diversity of channelopathies that can derail pain sensing. Indeed, an analysis of data from the UK Biobank, which has whole genome sequences and medical records for 100,000 Britons, revealed that voltage-gated sodium channels were the largest group of variants associated with neuropathic pain.
Based on his findings, Bennett advocates using both clinical testing data and gene sequencing to stratify patients according to which treatments are most likely to work for them. In particular, sodium channel blocking drugs appear to work much better in patients with variant channels predisposing them to pain.
Where does it hurt?
The meeting’s first regular session focused on efforts to dissect the central pain circuits in the nervous system. For Sarah Ross, the dissection is literal: she carefully removes a piece of a mouse spinal cord, along with the sensory nerves connected to a patch of skin from the animal’s hind paw, keeping all of the neuronal connections intact. Using luminescent probes, her team can then watch the activation of specific neurons in response to stimuli.
“We can see some neurons respond to heat, other neurons will respond to cool, other neurons will respond to mechanical stimuli,” said Ross.
Many neurons also respond to multiple stimuli, and mapping these responses reveals that distinct classes of neurons function as amplifiers, tuners, and integrators of pain signals.
Jing Wang studies what happens to pain signals in the cerebral cortex of the brain. Using optogenetics, which allows him to stimulate specific neurons in the brains of mice with light, he has identified subsets of neurons in the anterior cingulate cortex and prefrontal cortex that respond to pain.
In mice with experimentally induced chronic pain, low-intensity stimulation of the prefrontal cortex restores normal pain control. Wang’s lab is now studying ways to achieve similar responses with less invasive methods, including the drug ketamine and brain-machine interfaces.
“The cortex processes and regulates pain, but its normal endogenous function can be impaired by chronic pain, and [restoring cortical regulation] has the potential to transform pain treatment,” said Wang.
Part 2
Speakers
Aarno Palotie, MD, PhD Institute for Molecular Medicine, Finland
Luda Diatchenko, MD, PhD McGill University
Irene Tracey, MA (Oxon), DPhil, FRCA, FMedSci University of Oxford
Alban Latremoliere, MSc, PhD Johns Hopkins University
The pains of the father
Aarno Palotie began the meeting’s session on the genetics of pain by discussing his results from large-scale studies on migraine. With the exception of some rare, strictly inherited forms of the condition, these sporadic, debilitating headaches usually stem from variations in numerous common genes. To identify those genes, Palotie and a large team of collaborators scrutinized genetic and medical data from hundreds of thousands of migraine sufferers.
The effort revealed over 100 gene loci linked to migraine, mostly in regulatory regions associated with genes expressed in cardiovascular tissue and the central nervous system. Tracking those variants in another large data set revealed a cumulative effect.
“We can see that those with a high polygenic risk score, meaning a high load of common variants, they seem to have an earlier onset of migraine,” said Palotie.
Using data from the 500,000 participants in the UK Biobank, Luda Diatchenko and her colleagues have performed a similar analysis to identify genetic variants linked to chronic pain. The investigators subdivided chronic pain patients based on the type of pain they experienced, such as back pain, hip pain, knee pain, and multi-site pain.
Analyzing gene sequences for these sub-groups showed that multi-site pain had the highest correlation with specific gene variants. The gene most strongly linked to multi-site pain encodes a receptor protein involved in guiding nerve axons in development.
“This is one example of how [genome-wide association studies] can show us a new mechanism which contributes to human chronic pain conditions,” said Diatchenko.
On a scale of one to ten
The meeting’s third session focused on one of the biggest challenges in studying pain: measuring it. Clinical studies attempt to quantify pain severity with patient questionnaires, while animal experiments rely on behavioral responses, but both methods are notoriously unreliable.
Ilene Tracey hopes to solve that problem with neuroimaging, linking specific patterns of neuronal activation to painful stimuli.
“We’ve got now quite a good array of tools that are reasonably well developed and robust, that allow you to look at … ways that patients will experience their pain,” said Tracey.
By combining functional magnetic resonance imaging with electroencephalography, video analysis, and other sensing methods, this approach could allow researchers to quantify patient responses to pain treatment more reliably than current, fundamentally qualitative methods. Using machine learning, Tracey’s team can now measure pain and also distinguish different categories of it, such as physical versus emotional pain.
Sleep disturbances might also provide a pain gauge.
“The vast majority of patients with chronic pain suffer from poor sleep quality,” said Alban Latremoliere, who has been studying this connection as a potential pain biomarker.
By tracking electroencephalography and other measurements in sleeping mice, he and his colleagues have found that nerve injury, which causes chronic neuropathic pain, also changes the animals’ sleep architecture. Compared to uninjured animals, those with injured nerves suffer multiple brief interruptions in the non-REM phase of their sleep. When the injury heals, the normal sleep architecture returns; Latremoliere now hopes to use these patterns to quantify neuropathic pain severity and treatment efficacy in humans.
Part 3
Speakers
Greg Scherrer, PhD University of North Carolina
Venetia Zachariou, PhD, MBBS, MMed, MS Icahn School of Medicine at Mount Sinai
Rajesh Khanna, PhD New York University
David J. Julius, PhD University of California, San Francisco (UCSF)
The hurt blocker
As Greg Scherrer pointed out in the meeting’s fourth session, the real problem with pain isn’t that it exists, but that it’s unpleasant.
“If we were to understand how our brain collects this information from sensory neurons and the spinal cord to make pain unpleasant … maybe we’ll discover new ways to treat pain,” said Scherrer.
Indeed, a patient whose basolateral amygdala was removed to treat epilepsy could still sense painful stimuli, but didn’t label them as painful; the unpleasantness was gone. Examining mice with various alterations to the same brain region, Scherrer and his colleagues believe they have identified the amygdala cells responsible for connecting pain to unpleasantness. The investigators are now trying to identify receptors on those cells that would be good drug targets for new pain treatments.
Venetia Zachariou is also dissecting cellular signaling pathways to target in pain treatment, and her lab has uncovered several promising leads in recent years. When the COVID-19 pandemic derailed that work, though, the scientists quickly pivoted to apply their skills and techniques to study the new disease’s neuronal pathogenesis.
In a hamster model, they found that SARS-CoV-2, the virus that causes COVID-19, can acutely infect nerves in the dorsal root ganglia, which are also involved in pain sensing. Looking more closely at both the hamster model and a mouse model of SARS-CoV-2 infection, Zachariou has identified distinct changes in neurons’ gene expression patterns after virus infection, including a signature similar to that seen in models of neuropathic pain.
One of the most popular targets for researchers trying to develop new pain therapies is the sodium channel Nav1.7, a “pain amplifier” that several speakers at the meeting discussed. Rajesh Khanna is also interested in Nav1.7, but instead of targeting the protein directly, his team is trying to identify proteins that interact with it. That work led them to focus on collapsin response mediator protein 2 (Crmp2), which regulates Nav1.7 signaling.
Mice lacking Crmp2 are resistant to chronic pain, suggesting that drugs inhibiting its action would be good pain therapy candidates. After conducting extensive mechanistic studies, Khanna started a company to identify such inhibitors. So far, the company has optimized a lead compound that appears to stop chronic pain in animal models, without causing detectable side effects or tolerance.
You feel it in your gut
The meeting’s first day concluded with a keynote presentation by David Julius, who discussed his work on chronic visceral pain. This subtype of chronic pain, which can be caused by gut infection or non-infectious conditions such as inflammatory bowel disease, affects about 15% of the population. It’s three times more common in women than men, but nobody knows why.
“We’re interested in a particular aspect of visceral pain signaling, and that involves the interaction of sensory nerve fibers with the gut epithelium,” said Julius.
A subset of gut epithelial cells, called enterochromaffin cells, plays an outsize role in that interaction. Comprising only a fraction of a percentage of all gut epithelial cells, enterochromaffin cells make about 90% of the body’s serotonin, a potent neurotransmitter protein. They also fire electrical signals that could propagate to nearby neurons.
Julius wanted to analyze that process in live mice, but wasn’t happy with the standard mouse system for those types of experiments. That model involves putting irritants into a mouse’s gut to trigger a major inflammatory response, after which the animal remains hypersensitive to physical stimuli such as colon distention.
“Do we need to … put the mouse through all that, or can you have a model that’s simpler [and] does not require all the sequellae of an inflammatory episode?” asked Julius.
Instead, he and his colleagues first tried studying enterochromaffin cells in the context of cultured enteroids, pieces of intestinal epithelium that can mimic many aspects of gut biology in a petri dish. That system revealed that enterochromaffin cells respond to numerous compounds that fall into three general classes: ingested irritants, metabolites of common gut microbes, and endogenous regulatory hormones.
“So, we want to know how these cells integrate all this information, and what role this plays in maladaptive situations like [inflammatory bowel disease],” said Julius.
Based on those results, the researchers moved to a more complex system, an explanted piece of a mouse colon with its connecting nerves. Monitoring the electrical signals in the connected nerves reveals sensory signals from the explanted gut. In this setup, bathing the colon section with isovalerate, a bacterial metabolite that triggered a response from enterochromaffin cells in the enteroid experiment, makes it hypersensitive to subsequent physical or biochemical stimuli. This system also revealed different response patterns in guts from male and female mice.
Having demonstrated that isovalerate could induce gut hypersensitivity without the inflammatory response of harsher irritants, Julius’s team next tried looking at its effect in live mice. They used a small balloon in the colon, similar to an endoscope, as a stimulus, and monitored abdominal muscle contraction, a behavioral response to pain. Treating the mice with isovalerate increased the magnitude of subsequent pain responses potently in male mice, but less so in females, consistent with the explant results.
Subsequent experiments showed that enterochromaffin cells mediate these responses in live mice, apparently through both serotonin secretion and direct electrical signaling to neurons, and that these cells seem to respond differently in male and female mice.
Part 4
Speakers
Isaac Chiu, PhD Harvard Medical School
Camila Svensson, MS, PhD Karolinska Institutet
Alexander J. Davies, PhD Nuffield Department of Clinical Neurosciences
Dana Orange, MD Rockefeller University
Shrinivasan Raghuraman, PhD University of Utah
Jeffrey S. Mogil, PhD McGill University
Frank Porreca, PhD University of Arizona
Roger Fillingim, PhD University of Florida
Is antibody hurt?
Infections commonly cause pain, which researchers had long assumed was just a byproduct of the body’s inflammatory response. However, as Isaac Chiu explained in the meeting’s session on neuroimmune and autoimmune mechanisms in pain, infecting pathogens can also interact directly with nociceptors, or pain-sensing neurons. In one set of mouse experiments, for example, Chiu’s team found that nociceptors in the intestine can detect infection with Salmonella enterica, triggering a response that decreases the number of M cells, the specialized intestinal epithelial cells S. enterica preferentially infects.
“These neurons actually regulate cell numbers, [which] not only shuts down the number of gates for pathogen entry, it also helps a protective microbe … attach better on the surface of the epithelium,” said Chiu.
Camila Svensson discussed a pain condition that has baffled researchers and clinicians for decades: fibromyalgia. Characterized by pain hypersensitivity in soft tissues, sometimes coupled with neuropathic pain, the condition has long eluded efforts to uncover its etiology and underlying mechanisms.
After serendipitously discovering evidence for autoantibodies in fibromyalgia patients, Svensson has now developed human tissue and mouse models to characterize these antibodies in more detail. Transferring antibodies from fibromyalgia patients into mice causes pain hypersensitivity in the animals, and patients with higher levels of antibodies that react with human dorsal root ganglia cells have more severe disease.
“This suggests that there is an autoimmunity in subpopulations of fibromyalgia patients,” said Svensson, adding that besides suggesting a mechanism for the disease, autoantibody levels could help stratify patients in clinical trials.
The body’s own immune response is also a key contributor to chronic neuropathic pain, especially through neuroinflammation. Alexander Davies presented his work on another component of neuropathic pain: the cytotoxic cellular response.
Cytotoxic cells normally detect cancerous or virally-infected cells and target them for destruction, but they can also target injured neurons. Dissecting this response in an extensive series of experiments in mice, Davies and his colleagues have found that a specific receptor on cytotoxic cells allows them to target nociceptors after nerve injury, leading to degeneration of the damaged axons and resolution of pain hypersensitivity.
“So, our data suggest that intact sensory networks are a source of ongoing neuropathic hypersensitivity, and that by targeting those, we can help to resolve that,” said Davies.
Short, sharp shocks
Dana Orange gave the first of two short “data blitz” presentations, providing an overview of her group’s work on rheumatoid arthritis pain. Though inflammation of joints is a hallmark of this form of arthritis, Orange noticed an odd discrepancy.
“Patients who really don’t have a lot of inflammation were reporting a lot of pain,” she said.
Through a combination of human gene expression and mouse studies, she’s found that nerve development may play a bigger role than inflammation in driving rheumatoid arthritis pain.
Shrinivasan Raghuraman described his approach to characterizing chronic pain mechanisms, using a rat model. By collecting thousands of data points from individual rat neurons under different conditions, his lab has identified 19 different subsets of neurons with distinct responses to nerve injury. Raghuraman hopes that correlating the cells’ electrical responses with their gene transcription profiles will identify the underlying mechanisms driving chronic pain, and how different candidate drugs can influence it.
Sex and race
In the session on sex and ethnic differences in pain, Jeffrey Mogil began by pointing out a critical flaw in traditional pain research methods. Despite ample evidence that women experience more pain than men, “80 percent of preclinical studies use male rats or male mice only,” said Mogil.
That skew overlooks important differences in the biology of pain in males and females, though. In a mouse model of chronic neuropathic pain, for example, Mogil’s lab has linked chronic pain to premature shortening of chromosome ends, or telomeres – but only in male mice. Besides studying both sexes instead of just one, Mogil argued that researchers need to extend their animal studies to monitor chronic pain for longer time periods, to account for age-related phenomena such as telomere shortening.
Frank Porreca also looks at sex differences in pain, but focuses on the role of stress. Clinical data clearly show that stress exacerbates functional pain syndromes such as inflammatory bowel disease, migraine, and fibromyalgia, all of which are more prevalent in women than men.
To study such syndromes, Porreca’s team developed a mouse model in which they restrain the animals for a short time to induce stress, then treat them with a compound that causes headaches. These stress-primed mice develop allodynia, interpreting normally non-painful stimuli as painful, while controls that only got the headache-inducing compound didn’t.
While both male and female mice exhibited the same response, Porreca found that it operates through different biochemical mechanisms in the two sexes, underscoring the importance of studying both in preclinical research.
Unlike sex, race and ethnicity lack clear biological definitions.
“It’s important to keep in mind that race and ethnicity are not causal factors, but rather proxies for these many psychosocial and biopsychosocial factors, largely driven by systemic societal and environmental exposures,” said Roger Fillingim.
At the same time, the groups that suffer disproportionately from racial and ethnic health disparities are often the least-studied. That’s certainly the case in pain research and treatment. Indeed, experiments suggest that Black patients may experience more pain than white ones, but health data show they’re less likely to be treated for pain in hospitals and clinics.
Summarizing a large body of additional evidence for similar skews in various minoritized groups, Fillingim advocated more holistic approaches to pain research across and within sub-populations.
Part 5
Speakers
Alexander Chesler, PhD National Center for Complementary and Integrative Health (NCCIH), NIH
Patrik Ernfors, PhD Karolinska Institutet
Clifford Woolf, MD, PhD Harvard Medical School
Bryan Roth, MD, PhD University of North Carolina
Kelly Knopp, PhD Eli Lilly
Get the sensation
The meeting’s penultimate session focused on how sensory signals such as pain propagate toward the central nervous system. Alexander Chesler started the session with a discussion of his work on peripheral sensory neurons.
To study these cells, Chesler and his colleagues initially developed an elegant system that allowed them to probe the responses of individual mouse cells in the trigenimal ganglion, a nerve cluster that receives sensory signals. That revealed a specific subset of neurons that responded only to a painful stimulus, while other subsets responded to gentle touches. By extending the system with gene expression profiling, and correlating responses in the mouse with those in a human patient who lacks a receptor critical for mechanical sensation, the scientists are now tracing pain-sensing pathways in unprecedented detail.
Neurons aren’t the only cells carrying pain signals, though, as Patrik Ernfors has discovered. In tracing sensory circuits, he and his colleagues discovered that Schwann cells, support cells closely associated with peripheral neurons, are also stem cells that form a sensory organ under the skin.
Using genetically modified mouse models that allowed them to selectively activate these Schwann cells, Ernfors and his colleagues discovered that both the Schwann cells and their associated neurons can initiate acute pain sensations. Further work revealed that the Schwann cells also appear to become sensitized during the development of arthritis.
“We believe that we have found the mechanosensory skin organ that is associated with [mechanical pain sensation],” said Ernfors, adding that these cells could contribute to allodynia in arthritis.
Something for the pain
Clifford Woolf began the meeting’s final session, on finding new ways to treat pain, with a summary of his team’s novel approach to drug discovery. Currently, most pharmaceutical companies focus on finding compounds that can target specific cellular molecules known to be involved in pain, then trying to develop them into drugs.
In 2010, Woolf advocated an alternative strategy, screening drugs to find those that inhibit stem cell-derived pain-sensing neurons, without worrying about their mechanisms of action.
“However, the question was how to execute on this,” he said.
After extensive effort, his team can now derive the correct neuron types from patients’ cells. Screening libraries of compounds against these cells has yielded several promising hits, which inhibit pain signaling in nociceptors without affecting other cell types.
Others hope to broaden the scope of target-based drug screening, which has focused on a large and diverse class of cell surface proteins called G-protein coupled receptors, or GPCRs.
“But … when we mapped the drugs onto the phylogeny of all the [GPCRs] in the genome, only a few targets actually came out as being targets of approved drugs,” said Bryan Roth, adding that “there are many, many other potential targets for treating pain and other serious conditions.”
To test those targets, Roth’s team developed an assay that allows them to test drugs against a library encompassing 90% of GPCRs encoded in the human genome. That has revealed several new targets, which the researchers are now testing with more specific screens, ultimately hoping to develop safer opioids.
Kelly Knopp began the meeting’s final talk with the grim statistics of chronic pain: affecting about one fourth of the global population, the direct and indirect costs of this condition add up to more than a trillion dollars.
“[Meanwhile,] the probability of technical success for pain [drugs] is worse than any other therapeutic area,” said Knopp.
To address that, she and her colleagues have focused on establishing standardized protocols for phase 2 proof-of-concept trials of pain treatments. Their approach uses sophisticated statistical techniques and uniform trial designs to enable testing of many more drug candidates, without exceeding available funding and medical trial capacity.
After the presentations, a panel of speakers from the meeting discussed several of the field’s biggest challenges. Chief among them are the immense burden of opioid addiction, and the difficulty of shifting real-world clinical treatment toward less addictive but possibly less effective therapies for chronic pain. Despite the difficulties, many researchers in the field remain optimistic.
As Ilene Tracey said in her presentation, “We’re often quite doom and gloom in the pain field, [but] we’ve actually got a lot of different tools at our disposal, [and] we should be more confident about where the field has got to and where it can go quite rapidly.”
Although advances made in health and safety have more than doubled life expectancy throughout much of the world since 1900, it hasn’t been without consequence. Disease, disability, and frailty have all impacted the quality of life associated with these later years. This unfortunate reality was recently illuminated by the COVID-19 pandemic, which severely affected this population, likely due to physiological changes and preexisting conditions. Fortunately, a primary goal of geroscience researchers is to attenuate age-related health issues so that older people not only enjoy an improved quality of life, but also maintain the resilience to survive severe diseases and infections.
While it’s irrefutable that we cannot avoid aging, it’s no longer within the realm of science fiction for us to temper and even reverse the aging process. On May 19, 2021, the New York Academy of Sciences hosted a virtual symposium that brought together geroscience experts spanning various disciplines, including genetics, endocrinology, gerontology, clinical psychology, and more. Speakers discussed targeting the key hallmarks of aging, developing biomarkers for geriatric therapies, and translating findings that extend healthspan and lifespan to the clinic.
Symposium Highlights
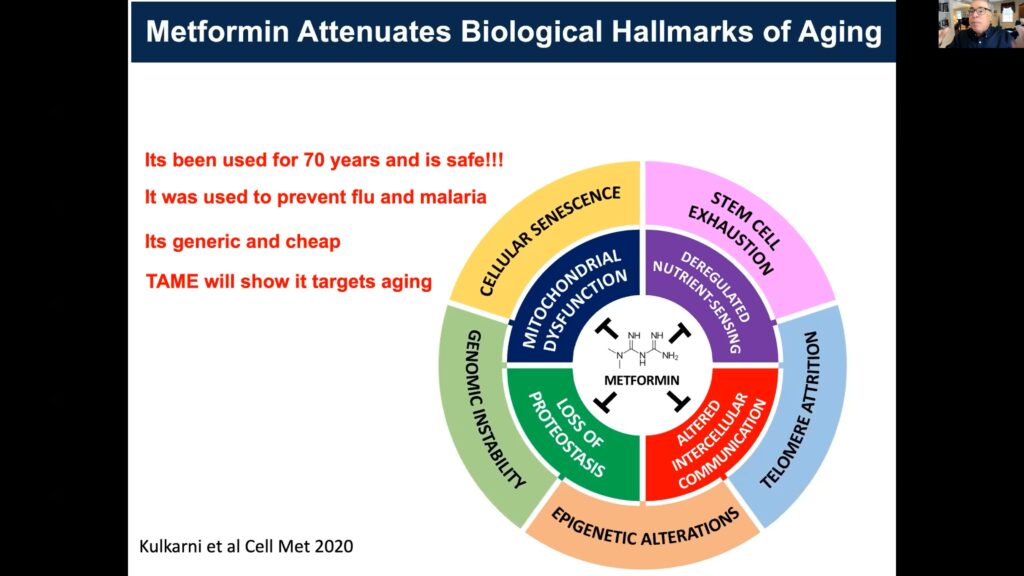
The Target Aging with Metformin study uses the FDA approved anti-diabetic metformin, which targets the hallmarks of aging, to investigate the prevention of age-related diseases.
Precluding the age-associated decline of chaperon-mediated autophagy restrains the aggregating effects of Alzheimer’s disease and extends lifespan in murine models.
Lower IGF-1 levels in older adults are associated with decreased cognitive impairment, age-related diseases, and mortality.
Epigenetic clocks can be applied to study biological aging differences, with accelerated epigenetic aging correlating with the prevalence and incidence of morbidity and mortality.
The metabolome is a powerful locus of opportunity to bridge the gap between genotype and age.
Alternative splicing is upregulated in response to declining mitochondrial function and increasing age.
Senescent cells upregulate pro-survival pathways, and their elimination alleviates diverse age-related conditions.
The mitochondrial-derived peptides humanin and MOTS-c are associated with increased longevity in animal models and humans.
Speakers
Nir Barzilai, MD Albert Einstein College of Medicine
Ana Maria Cuervo, MD, PhD Albert Einstein College of Medicine
Sofiya Milman, MD Albert Einstein College of Medicine
Morgan Levine, PhD Yale School of Medicine
Daniel Promislow, PhD University of Washington
Luigi Ferrucci, MD, PhD National Institute on Aging, National Institutes of Health
James Kirkland, MD, PhD Mayo Clinic
Pinchas Cohen, MD USC Leonard Davis School of Gerontology
Targetable Aging Processes
Speakers
Nir Barzilai, MD Albert Einstein College of Medicine
Ana Maria Cuervo, MD, PhD Albert Einstein College of Medicine
Keynote: Age Later: Translational Geroscience
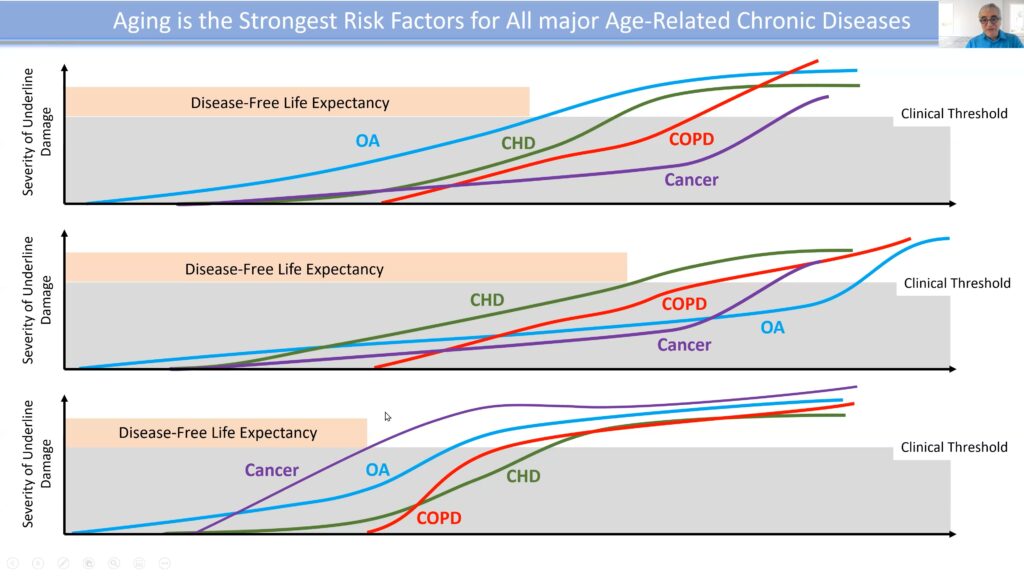
Aging is the strongest risk factor for all age-related diseases, with diverse maladies accumulating during the later years of life. Hence, to abate or avert the relevant disorders, it’s critical to target the central driver—aging itself. Physician Nir Barzilai, the founding director of the Institute for Aging Research, investigates the genetics of longevity by studying centenarians and their offspring, interrogating the hypothesis that these individuals have genes that prolong aging and protect against age-related diseases.
Using Slow Off-Rate Modified Aptamer, Barzilai’s team assessed 5,000 proteins in a population of 1,000 individuals between the ages of 65-95, a period during which aging accelerates. Results demonstrated a significant change in the level of hundreds of proteins as a function of age. Among the top hits were proteins from collagen breakdown of tissue and cellular products, highlighting the pivotal role this process plays in aging, and suggesting that deterring disintegration may be a universal biomarker for geroprotection.
Metformin, a long-standing FDA approved anti-diabetic, targets the complement of aging indications.
A predominant challenge to translating advances made in geroscience from animal models to humans is the FDA, which currently doesn’t consider aging a disease indication or preventable condition. Barzilai and others are utilizing metformin, an FDA-approved anti-diabetic, to refute this contention. Various groups have shown that metformin has substantial effects on human healthspan, including delaying type-2 diabetes mellitus (T2DM). In this patient subset, metformin also impedes cardiovascular disease, cognitive decline, and Alzheimer’s and is associated with decreased cancer incidence, with population effects approaching 30% in all cases.
Barzilai’s team designed the Target Aging with Metformin, or TAME, study to investigate whether or not there’s a shift in the timeline of disease occurrence between a cohort receiving metformin versus a control cohort. Various biomarkers of aging and age-related diseases will be used to provide convergent evidence of broad, age-related effects, while also establishing a resource for innovation and discovery of emergent biomarkers.
“The most important thing for us is to develop biomarkers that will change when we use a gerotherapuetic,” Barzilai asserted, as this will expedite therapeutic prospects.
Targeting Selective Autophagy in Aging and Age-related Diseases
Physician-scientist Ana Maria Cuervo’s research seeks to understand the molecular basis of autophagy dysfunction with age and the contribution of defects in this cellular pathway to diseases such as neurodegeneration, metabolic disorders, and cancer. Autophagy belongs to the proteostasis network, which regulates protein content and quality control.
Chaperon-mediated autophagy (CMA) is a subset of the mammalian autophagy program that directly targets proteins to the lysosome for degradation. CMA has been shown to decrease with age in human and animal models. Cuervo’s lab developed a fluorescent murine reporter construct to visualize CMA and track the kinetics of its activity in different organs.
Blocking this pathway in neurons resulted in the aggregation of proteins like α-synuclein (α-syn), tau, and others that are causal in Alzheimer’s Disease (AD). Additionally, CMA reporter mice crossed with a mouse model of AD revealed that CMA activity dramatically decreases in the neurons of AD mice.
Leveraging these findings, Cuervo’s group generated a mouse model to restore CMA activity conditionally. Mice with preserved CMA exhibited an extended median and maximal lifespan compared to controls. Evaluation of the proteostasis network in mice with and without CMA restoration revealed major changes in the proteome. Mice in which CMA was preserved more closely resembled younger animals than their age-matched controls.
“By acting in one of these pathways, we can have an impact in the other hallmarks of aging… because of this interconnection among [them],” Cuervo emphasized.
A compound to selectively activate CMA was developed and tested in an AD model, with results illustrating a reduction in tau pathology and microglial activation in the presence of this agent.
Sofiya Milman, MD Albert Einstein College of Medicine
Morgan Levine, PhD Yale School of Medicine
Translational Geroscience: Role of IGF-1 in Human Healthspan and Lifespan
Physician Sofiya Milman conducts translational research to uncover the genomic mechanisms regulating the endocrine and metabolic pathways involved in age-related conditions like diabetes, cardiovascular disorders, and Alzheimer’s.
“The goal of geroscience is really to extend healthspan, and not necessarily lifespan,” Milman opened. “What we’re really trying to do is to compress the period of morbidity.”
To discover the biological pathways that allow humans to live long, healthy lives, Milman’s team focused on IGF-1: a reduction of this factor has been consistently shown to extend healthspan and lifespan in models. IGF-1 levels peak during the teenage years before gradually declining. If the reduction of IGF-1 protects from aging, Milman reasoned that lower IGF-1 levels would delay aging and prevent age-related diseases.
Examining a cohort of centenarians expressing lower levels of IGF-1 revealed a 50% reduction in cognitive impairment compared to higher IGF-1 level controls. Genetic studies demonstrated that centenarians were enriched for rare mutations in the IGF-1 receptor that diminished signaling. Additionally, individuals 65+ with low IGF-1 had less cognitive impairment, and delayed onset of cognitive impairment, multi-morbidities, and mortality.
Milman’s team also addressed the link between IGF-1 and age. Younger individuals with lower levels of IGF-1 were at an increased risk for mortality and age-related diseases compared to older individuals, while higher levels of IGF-1 in older adults were associated with increased risk. This suggests that the IGF-1 network aligns with the concept of antagonistic pleiotropy, wherein a factor that’s beneficial to individuals when they’re younger may become harmful when they’re older. It’s advantageous to maintain functionality of proteostasis and resilience as an individual gets older, but IFG-1 inhibits programs involved in these processes.
“So from this, we think it would be wise to maintain IGF-1 levels in youth, but to reduce them with aging,” Milman concluded.
Epigenetic Biomarker of Aging for Lifespan and Healthspan
Biological age is defined by changes or alterations in a living system that renders it more vulnerable to failure and is behind the age-related increase in susceptibility to chronic diseases. Unlike chronological age, it is very difficult to measure because it’s unobservable.
Morgan Levine integrates theories and methods from statistical genetics, computational biology, and mathematical demography to develop biomarkers of aging for humans and animal models. Among this work are efforts to establish systems-level outcome measures of aging to facilitate evaluation for gero-protective interventions.
“There’s some disagreement on how we actually quantify [biological age],” Levine started. “But I would argue that it’s really important to try and do so, because quantifying [this] will really help us in a number of endeavors in the field.”
Levin’s lab is particularly interested in epigenetic aging, as aging drastically remodels the DNA methylation landscape, with widespread increases and decreases as a function of age.
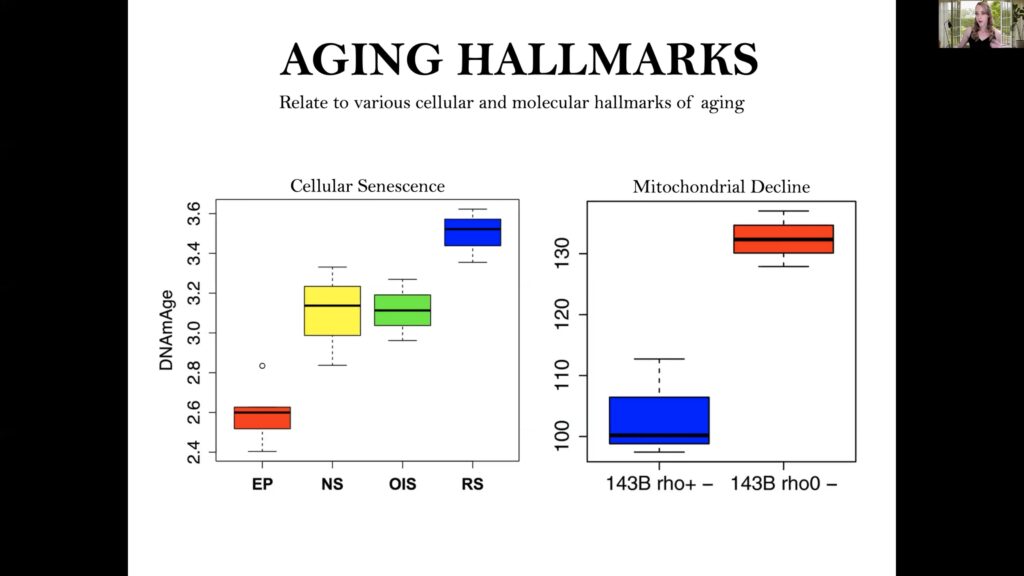
Senescent cells and cells with disrupted energy production show accelerated epigenetic aging.
Epigenetic clocks estimate DNA methylation across the genome and combine supervised machine-learning approaches to develop predictors of biological age.
“We think people who have a predicted [epigenetic] age that’s younger than their chronological age should be actually aging slower, whereas the opposite is true for people that have a genetic age that is predicted higher,” said Levine.
Applying these measures to diseased states yielded several pertinent findings. For example, individuals who have pathologically diagnosed Alzheimer’s post-mortem show accelerated epigenetic aging in their brain relative to their chronological age. Tissue differences were also captured, revealing that tissues seem to age asynchronously, with highly proliferative tissues and tumor cells having accelerated aging compared to slower aging brain tissue.
Levine’s group also evaluated cellular senescence and energy disruption, with results revealing that near senescent, HRAS oncogene induced senescent, and replicative stress senescent cells have an acceleration in epigenetic age compared to early parental control cells. Additionally, deletion of mitochondrial DNA accelerated epigenetic aging, while caloric restriction in mice stalled their epigenetic clocks.
Luigi Ferrucci National Institute on Aging, National Institutes of Health
Metabolomics in the Search for Biomarkers and Mechanisms of Aging
Daniel Promislow applies metabolomics and systems biology approaches to study aging, with a focus on understanding the evolutionary and molecular traits that shape fitness in the natural human population. Although genome-wide association studies have allowed researchers to identify thousands of polymorphisms associated with the complement of measurable traits, including aging, the disparities identified explain less than half of 1% of the phenotypic variations.
Many genes interacting with each other ultimately influence phenotypes, and the biological distance between the two is astronomical. To bridge this gap, researchers use endophenotypes—from the epigenome, transcriptome, proteome, metabolome, and microbiome—along with various omics approaches. Promislow’s lab focuses on the metabolome, which integrates information from the environment and genotype to ultimately affect aging.
Promislow’s team utilizes translational metabolomics in various insect and animal models to understand and translate aging patterns to human populations. Applying this approach to Drosophila demonstrated that the metabolome could predict stress resistance, completely separating groups of sensitive or resistant flies to a metabolic stressor by principal component analysis. These effects could not be recapitulated with a whole fly genome sequence dataset. Evaluating response to diet restriction (DR) also revealed changes in metabolite levels with age. Among nearly 200 different inbred strains, roughly 75% showed a benefit to DR.
“Interestingly, the effect of specific genetic variants on the lifespan response was very weak,” Promislow began. “But we did find genes that were associated with metabolites, which were associated with the lifespan response, reinforcing this idea…that the metabolite profile can be a kind of bridge between genotype and phenotype.”
Promislow’s group also demonstrated that the metabolome could serve as a biological clock, revealing that shorter-lived genotypes appeared to have a higher biological age than expected for their chronological age.
Translational Potential of the Biology of Aging
As individuals age, the incidence of chronic disease increase, and disease progression quickens. Physician-scientist Luigi Ferrucci aims to interrogate the causal pathways that lead to progressive physical and cognitive decline in aging.
Cellular damage is accumulated during a person’s life, eventually reaching a pathology threshold that becomes clinically relevant when the damage presents as a disorder. Conventionally, the disease is often only addressed once it reaches this stage. The problem with this approach is that the present disease is often a marker of a more profound and invasive disorder to come.
“[Instead], we need to measure the underlying force that determines the emergence of diseases and their consequences,” Ferrucci argued.
By interfering with the basic mechanisms of aging to curtail it, broader effects of abating multiple chronic disorders can be achieved.
Cellular damage is accumulated over the course of an individual’s lifetime, with disease presenting once the clinical threshold for a given disorder is reached.
The rate of biological aging can be defined by the ratio of cellular damage accumulation to repair capacity. If the rate of damage accretion is fast, but the repair capacity is high, there won’t be an accumulation of damage, and aging will be slowed. However, when damage outpaces repair, aging accelerates.
Repair pathways require energy to operate effectively, and mitochondrial function declines dramatically with age. Ferrucci’s team discovered that this decline is associated with an upregulation of alternative splicing of mitochondrial proteins. Delving deeper into this mechanism, they applied gene set enrichment analysis to 5,325 RNAs with at least one splice variant significantly altered in response to changing mitochondrial function, as measured by AMPK and aging.
Among the top hits were GLUT4, VEGFA, IRS2, mTOR, PI3K, ULK1, ACC1, NRF2, and PGC1-α. Of note, the splice A variant of the topmost hit, VEGFA, appeared to be geronic, while the B variant appeared to be anti-geronic, with the ratio of these variants declining with age. Thus, alternative splicing is a method by which the body copes with energy decline due to mitochondrial dysfunction.
Translational Research for Healthspan and Lifespan
Speakers
Pat Furlong, Panelist Parent Project Muscular Distrophy
Roman J. Giger University of Michigan School of Medicine
Senolytics: The Path to Translation
Physician-scientist James Kirkland studies the impact of cellular aging, specifically senescence, on age-related dysfunction and chronic diseases to develop methods for removing these cells and attenuating their deleterious effects. Senescent cells accumulate with aging and diseases, eliminating cells around them due to their senescence-associated secretory phenotype (SASP), which 30%-70% of senescent cells exhibit under most conditions.
Kirkland’s team applied a bioinformatics-based approach to analyze SASP proteomic databases, revealing that pro-survival networks are upregulated, with diverse senescent cells relying on different pathways. Several agents, termed senolytics, were identified that could target multiple nodes of these cascades.
“We’re moving away from the one drug, one target, one disease approach here,” said Kirkland, “to try and use agents that have multiple targets, or combinations of agents, to go after networks, and to go after senescent cells by doing this, and thereby improve…multiple conditions.”
Dasatinib (D), a SRC kinase inhibitor, preferentially killed senescent preadipocytes, which relied on survival pathways that signal through this kinase. Quercetin (Q) eliminated senescent human umbilical endothelial cells (HUVECs), which partly act through the Bcl-2 family and others that this cell type is susceptible to.
In an in vivo experiment, combining Dasatinib with Quercetin (D+Q) cleared transplanted luciferase-expressing senescent preadipocytes from mice, explicitly targeting those cells with a SASP. A single dose of senolytics also alleviated radiation-induced gait disturbance in mice, with the effects persisting long-term. Bi-weekly dosing reduced physical dysfunction in older mice, as measured by parameters of maximal speed, including treadmill and hanging endurance, grip strength, and daily activity, with D+Q significantly increasing performance across the board.
Many conditions have now been shown to be alleviated by various senolytics in a range of mouse models, with D+Q delaying death from all causes, and increasing healthspan and median lifespan.
Keynote: Mitochondrial-derived Peptides (MDPs) and the Regulation of Aging Processes
The discovery of mitochondrial peptides (MDPs), encoded from small genes less than 100 codons in length, established the birth and advancement of the microprotein subfield. Physician Pinchas Cohen works to understand mitochondrial biology and characterize MDPs, exploiting findings to target aging. MDPs are secreted from cells and circulate within the body.
“Overall, they serve as protective factors, or hormones if you will, that act in the brain, the heart, the liver, the muscle, and other organs,” Cohen stated.
Among these MDPs, Cohen’s lab identified humanin, encoded from the 16S region of mtDNA, and MOTS-c, encoded from the 12S region.
Humanin has a strong protective effect on neurons and against atherosclerosis, mitigates the side effects of chemotherapy while enhancing its benefit, and is related to longevity in model organisms and humans. Cohen’s lab employs mitochondrial-wide association studies (MiWAS) to link the dysfunction of MDPs to disease. MiWAS identified a single-nucleotide polymorphism (SNPs) in the humanin gene (rs2854128) associated with reduced levels and cognitive decline in humans and mice. Supplementing humanin in mice carrying this SNP improved their cognition.
MOTS-c is a novel exercise mimetic that has potential utility in numerous age-related diseases. Mice on a high fat diet receiving MOTS-c had dramatically lower weight compared to controls. MOTS-c treatment also improved exercise tolerance and performance in middle-aged and old mice, with older mice displaying the most dramatic improvement.
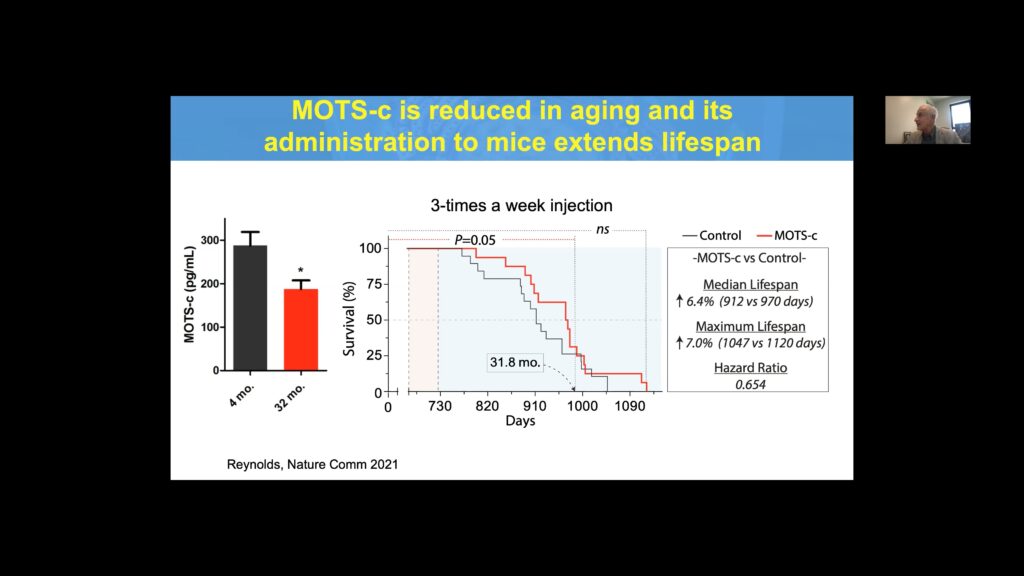
MOTS-c levels are diminished in older mice, and supplementation of MOTS-c in this cohort increases both median and maximum lifespan compared to controls.
Cohen’s group also identified a link between a SNP in MOTS-c–K14Q–which nullifies MOTS-c activity and the risk of diabetes in males of the Asian population. Evaluating Japanese males from three cohorts revealed a 50% increase in the risk of diabetes for carriers, with almost double the risk seen exclusively in men who were sedentary. Like other MDPs, MOTS-c is reduced with age, and its administration to mice significantly extends lifespan.
“I think that everything we do in the aging field can be reduced to trying to simulate the beneficial effects of a healthy lifestyle, particularly diet…and exercise,” Cohen said. “We think that…mitochondria are the main source of action [here] by inducing the production of peptides such as MOTS-c, humanin, and others.”
Currently the FDA categorizes psychedelics such as LSD and psilocybin as Schedule I drugs, indicating that these substances have no medical value. Despite this classification, a resurgence of research in approved labs has demonstrated therapeutic benefits of psychedelics for treatment of psychiatric disorders.
Of note, a recent trial on the effects of MDMA-assisted therapy for post-traumatic stress disorder (PTSD) showed a reduction in the severity of patient symptoms compared with the placebo arm of the trial, providing hope for the future approval of MDMA for therapeutic use. The exciting findings from this study as well as and investigations into other psychedelics are instigating a paradigm shift for treatment-resistant psychiatric conditions, along with increased public interest and efforts to legalize psychedelics for medicinal use.
The New York Academy of Sciences hosted a panel discussion bringing together leading scientists in the fields of pharmacology, neuroscience, and psychiatry to discuss how psychedelics work in the brain to produce therapeutic benefits for depression and other mood disorders. The conversation commenced a description of the socio-political context of psychedelics research, spanning the rise of psychedelics research in the 1950s, restrictions in the 1960s, renewed interest in the 1990s, and present day clinical trials for patients with depression and various other mood disorders.
The program continued by spotlighting the different types of classical and non-traditional psychedelics that are currently being investigated (e.g., psilocybin, MDMA, and ketamine) and how they work to produce therapeutic effects. Panelists concluded the conversation by sharing insights into the use of psychedelics in treatment settings, including explaining the process of facilitated treatment and the role of the therapist/guide during the psychedelic experience (including preparatory therapy, peak effects, and integration).
In this eBriefing, you will learn:
The socio-political history of psychedelic research for human health
The difference between classic and non-traditional psychedelics
The effects of psychedelics on the brain and targets
The role of the hallucinogenic experience
The role of psychological support during the psychedelic experience
Psychedelics for the Treatment of Depression and Psychiatric Disorders
Moderator
John Krystal, MD Yale School of Medicine
Speakers
Roland Griffiths, PhD Johns Hopkins University School of Medicine
David E. Nichols, PhD Heffter Research Institute
Rachel Yehuda, PhD Icahn School of Medicine at Mt. Sinai
John Krystal, MD Yale School of Medicine
Dr. John Krystal is the Robert L. McNeil, Jr., Professor of Translational Research; Professor of Psychiatry, Neuroscience, and Psychology; and Chair of the Department of Psychiatry at the Yale University. He is also Chief of Psychiatry and Behavioral Health at Yale-New Haven Hospital. He is a graduate of the University of Chicago, Yale University School of Medicine, and the Yale Psychiatry Residency Training Program.
Dr. Krystal has published extensively on the neurobiology and treatment of schizophrenia, alcoholism, PTSD, and depression. Notably, his laboratory discovered the rapid antidepressant effects of ketamine in humans. He is the Director of the NIAAA Center for the Translational Neuroscience of Alcoholism and the Clinical Neuroscience Division of the VA National Center for PTSD. Dr. Krystal is a member of the U.S. National Academy of Medicine and a Fellow of the American Association for the Advancement of Science. Currently, he is co-director of the Neuroscience Forum of the U.S. National Academies of Sciences, Engineering, and Medicine; and editor of Biological Psychiatry (IF=12.1).
He has chaired the NIMH Board of Scientific Counselors and served on the national advisory councils for both NIMH and NIAAA. Also, he is past president of the American College of Neuropsychopharmacology (ACNP) and International College of Neuropsychopharmacology (CINP).
Roland Griffiths, PhD Johns Hopkins University School of Medicine
Roland Griffiths is Professor in the Departments of Psychiatry and Neurosciences and Director of the Center for Psychedelic and Consciousness Research at the Johns Hopkins University School of Medicine. His principal research focus in both clinical and preclinical laboratories has been on the behavioral and subjective effects of mood-altering drugs and he is author of over 400 scientific publications. He has conducted extensive research with sedative-hypnotics, caffeine, and novel mood-altering drugs.
About 20 years ago, he initiated a research program at Johns Hopkins investigating effects of the classic psychedelic substance psilocybin, the active component in “magic mushrooms.” Remarkably, many research participants rate their experience of psilocybin as among the most personally meaningful of their lives, and they attribute enduring positive changes in moods, attitudes and behavior months to years after the experience. Completed and ongoing studies include those in healthy volunteers, in beginning and long-term meditators, and in religious leaders.
Therapeutic studies with psilocybin include treatment of psychological distress in cancer patients, major depressive disorder, nicotine addiction, anorexia nervosa, and various other psychiatric disorders. Related studies of brain imaging and drug interactions are examining pharmacological and neural mechanisms of action. His research group has also conducted a series of survey studies characterizing various naturally-occurring and psychedelic-occasioned transformative experiences including mystical experiences, entity and God-encounter experiences, Near Death experiences, and experiences claimed to reduce depression, anxiety, and substance use disorders.
David E. Nichols, PhD Heffter Research Institute
David E. Nichols previously held the Robert C. and Charlotte P. Anderson Distinguished Chair in Pharmacology and in addition was a Distinguished Professor of Medicinal Chemistry and Molecular Pharmacology at the Purdue University College of Pharmacy. He was continuously funded by the NIH for nearly three decades and served on numerous government review panels. His two principal research areas focused on drugs that affect serotonin and dopamine transmission in the CNS.
He began medicinal chemistry research on hallucinogens in 1969 and has been internationally recognized as a top expert on the medicinal chemistry of psychedelics (hallucinogens). He has published more than 300 scientific articles, book chapters, and monographs. In 1993 he founded the Heffter Research Institute, which has supported and funded clinical research with psilocybin and led the so-called “renaissance in psychedelic research.”
Rachel Yehuda, PhD Icahn School of Medicine at Mt. Sinai
Rachel Yehuda, Ph.D. is the Director of the Center for the Study of Psychedelic Psychotherapy and Trauma, Vice Chair for Veterans Affairs for the Psychiatry Department and a Professor of Psychiatry and Neuroscience at the Icahn School of Medicine at Mount Sinai as well as the Director of Mental Health at the Bronx Veterans Affairs Medical Center and the Director of the Traumatic Stress Studies Division.
Throughout her career her research has focused on the study of the enduring effects of trauma exposure, particularly PTSD, as well as associations between biological and psychological measures. She has investigated novel treatment approaches for PTSD and the biological factors that may contribute to differing treatment outcomes for the purpose of developing personalized medicine strategies for treatment matching in PTSD. This work has resulted in an approved US patent for a PTSD blood test.
Recently, Dr. Yehuda’s laboratory has used advances in stem cell technology to examine PTSD gene expression networks in induced neurons. The Center for Psychedelic Psychotherapy and Trauma integrates sophisticated brain imaging and molecular neuroscience in PTSD with clinical trials using MDMA assisted psychotherapy and other related medicines. She has authored more than 450 published papers, chapters, and books in the field of trauma and resilience, focusing on topics such as PTSD prevention and treatment, molecular biomarkers of stress vulnerability and resilience, and intergenerational effects of trauma and PTSD.
According to the National Center for Education Statistics, white males made up 53% of all full-time professors in 2018. And while the “STEM pipeline” is becoming more diverse–more than 40% of women and roughly 15% of people of color receive their PhDs in STEM fields–colleges and universities need to implement inclusive policies to initiate change on a large scale.
On October 9, 2020, the New York Academy of Sciences hosted a webinar with Georgetown University Medical Center affiliates to share their progressive efforts to decrease systemic inequities and improve workplace culture at their institution. In 2019, the university launched the Bias Reduction and Improvement Coaching (BRIC) program to raise awareness of unconscious bias and attenuate systemic barriers at institutions with the hope of promoting diversity and inclusion in STEM.
Highlights
Bias impacts application, hiring, and promotion processes, as people make decisions based on shortcuts, unconscious preferences, and assumptions.
The Bias Reduction and Improvement Coaching (BRIC) program brings together a group of individuals from various demographic backgrounds for training in the skills and language needed to raise awareness of bias.
This “train the trainer” model empowers people to feel confident starting conversations about prejudice and how to mitigate bias in their respective departments and workplaces.
Speakers
Susan Cheng, EdLD, MPP Georgetown University Medical Center
Kristi Graves, PhD Georgetown University Medical Center
Caleb McKinney, PhD, MPS Georgetown University Medical Center
Reducing Systemic Inequities in Academia
Unconscious Bias in STEM
Search committees looking to fill a job should be as objective as possible, especially when studies have shown that teams made up of diverse people are more innovative and high-performing. However, people rely on mental shortcuts and assumptions when making hiring decisions. They often use reflexive habits and exhibit unconscious preferences without realizing it.
Caleb McKinney, who trained as a microbiologist, transitioned to science education, and is now an Assistant Professor and Assistant Dean for Graduate and Postdoctoral Training and Development, related this phenomenon of reflexive habits to a “hot pot.” You learn from previous experience to pull your hand away when a stove is hot. With the same mindset, you can use your prior knowledge to make quick assumptions and form preferences about someone. He urged everyone to take an Implicit Association Test online to learn more about unconscious bias.
But how does conscious and unconscious bias impact STEM community development? Assistant Professor and Senior Associate Dean for Diversity, Equity, and Inclusion, Susan Cheng, noted one example. STEM emphasizes innate intelligence over hard work, but in letters of recommendation, professors are more likely to refer to male scientists as “brilliant,” whereas female scientists are “productive.” The way a job description is written says a lot about what admissions may be looking for in a student or what faculty may desire during recruitment. Search committees may deem a person “not a good fit” for the institution. The only way to combat this is to use checkboxes to ensure job description criteria are followed systematically. “Implicit biases are always in the background, and you need to manage them actively,” said Cheng.
Kristi Graves, a clinical health psychologist and Associate Professor of Oncology, explained that bias also affects STEM professionals’ upward trajectories. For instance, scholarly productivity metrics are very numeric and usually include the number of papers published, impact factor for the journal in which you’ve published, and the amount of grant funding you’ve obtained. But faculty members don’t have access to the same opportunities. A male professor going to another male for a collaboration (because he is like him) is an example of similarity bias.
It’s critical to note that Black, Indigenous, People of Color (BIPOC) make up a small percentage of faculty members. BIPOC faculty members often act as the representative BIPOC for diversity panels and mentoring groups, which takes time away from work and research. And the amount of time spent on essential work and research affects prospects for promotions. Graves believes that hiring committees should have explicit discussions about implicit bias throughout the year to increase faculty diversity. “Everyone has bias,” said Graves. “The trick is to try to become aware of the bias, and then when you notice it, you do something about it so the negative impact that flows from that bias is not sustained or perpetuated.”
Although many colleges and universities have increased awareness and implemented more inclusive policies, the culture has not shifted enough to facilitate a more diverse institutional community. Even representative images on posters and brochures should indicate that a university values different types of people in STEM and that the depicted individuals can serve as role models for scientists who want to know what the institution values.
Understanding the BRIC Program
All three panelists have been heavily involved in the Bias Reduction and Improvement Coaching (BRIC) program at Georgetown University. The program leaders selected people from different backgrounds in various departments across the university for the program. Supervisor or departmental approval was required to participate since the program would take away hours spent on “numeric success metrics.” The inaugural group of 27 coaches—five of whom are coach leads—went through four, three-hour immersive sessions held quarterly. During these meetings, which covered the science of bias and its impact on hiring, promotions, retention, and overall workplace culture, participants learned evidence-based strategies to raise awareness and reduce discrimination. Participants led presentations and department talks on what they learned and received feedback from the coach leads.
The program’s goal is to have many people who can confidently initiate conversations about bias in their workplace. It is designed to establish training across the medical center by providing a faculty learning environment, explained Cheng. The messenger is so important because having the information come from a colleague you know and trust to understand the institutional context you work in is invaluable.
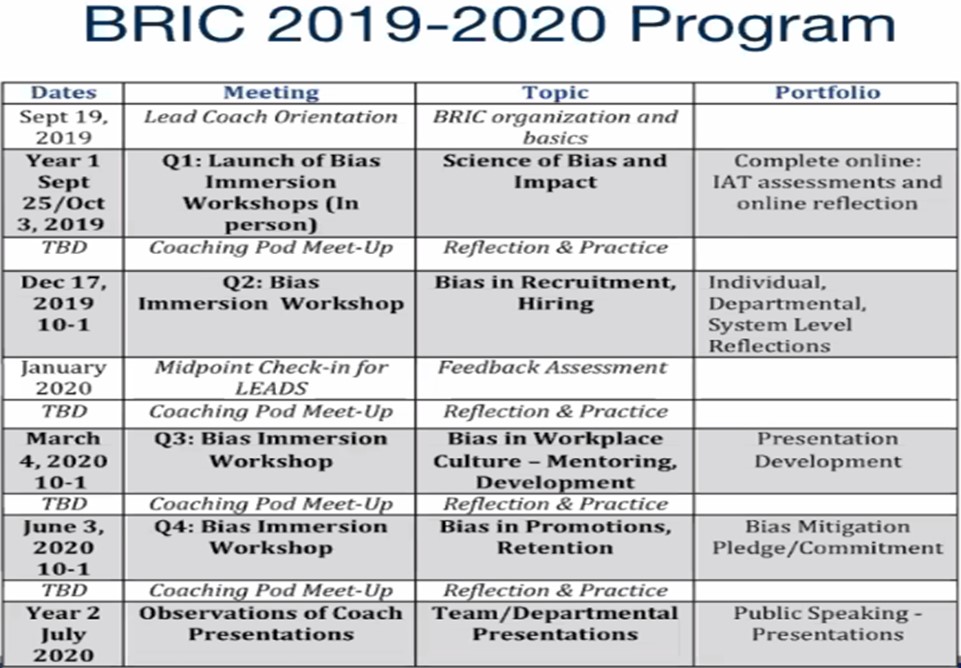
Figure 1. The BRIC program schedule included three-hour sessions, with smaller pod meetings in between, and presentation feedback for the participants before disseminating the information to colleagues.
McKinney participated in the BRIC program and pinpointed three main attributes of the initiative. The training provided self-knowledge to reflect upon one’s personal bias, leadership skills to feel equipped to speak up about bias when necessary, and the ability to communicate these strategies when training others. Participants were asked to reflect on their time in the program and took surveys to assess its impact. Additionally, audience members from BRIC coaches’ presentations were surveyed to see if it was scaled to the department, and follow-ups were conducted to see if departments made any significant changes.
Program Outcomes
Although the initial training was geared toward faculty and staff, post-docs and graduate students organized bias reduction workshops and helped create presentations for their departments. Cheng believes that many students have already developed these skills, while faculty and staff are trying to catch up by participating in the available programs. In addition, departments are asking for workshops on microaggressions, anti-racism, and bias. Graves explained that the program built tremendous confidence for those presenting the material in a safe and confidential space. She also shared a reflection from a participant in the program who felt BRIC was “an effective approach to raise awareness about unconscious beliefs and attitudes and to discover biases in a non-confrontational manner.”
By bringing in participants from different sections of the medical center, the BRIC program facilitated collaboration between faculty and staff from various departments who would not typically work together. This teamwork increased cognitive diversity and allowed participants to build on each other’s thoughts in a broad group discussion. McKinney emphasized that BRIC fostered peer mentorship and feedback, and helped strengthen a sense of community throughout Georgetown University Medical Center.
He was impressed with how much he learned about himself while participating in the program. “To be truly empathetic, you have to be self-aware. You have to recognize and challenge the assumptions you may have about people and situations,” McKinney said. “The BRIC programs allows you to put yourself in other people’s shoes. You learn from each other how people’s experiences and backgrounds shape their individual context, and that’s so important for building empathy.”
Metrics for Success
The panelists shared an essential set of standards that needed to be met for the BRIC program to be successful. Most importantly, improving diversity requires commitment from everyone. That commitment starts with raising awareness and then addressing the issues to create a sense of belonging in the workplace. “Once people are more aware of these biases and start to engage in mechanisms to reduce those biases, you can really [assess an] environment that’s hostile and not welcoming,” said Cheng.
Graves sought feedback from leadership at Georgetown before starting the program. “Until you enact specific behaviors and policy changes, it doesn’t mean a lot,” she said. The institution is responsible for creating an inclusive environment for trainees, post-docs, and faculty and ensuring that people have equal opportunities to succeed. Establishing an inclusive environment doesn’t have to be a top-down method; valuable feedback can come from anyone.
Cheng also highlighted the importance of decisions and transparency. Critical decision making, which includes communicating news and defining expectations and limitations, should be collaborative. For example, department chairs should define a clear set of expectations, goals, and values before a selection process, and the selection committee should routinely review those criteria. “In STEM, we should be really good at creating objective definitions of how we know when we’ve made our goal,” said Graves.
In addition, all employees should periodically revisit training on inclusion and bias, not just at the onset of the job. McKinney advocated for structured mentorship for minority groups, including cross-cultural mentoring relationships. “Mentors from majority backgrounds have an opportunity to shape retention by fostering these welcoming environments for junior individuals to succeed,” he said. Mentorship also includes facilitating career identities for graduate students and post-docs. While it is critical to have support and map out your trajectory on an academic career path, mentors can also highlight opportunities in industry and focus on broad career paths.
The Future of BRIC
Bias awareness is critical now, when many interviews and meetings are being held virtually due to the pandemic. Graves saw this as a positive because it allows institutions to create new networks and reach students early on in their academic trajectories. They can reach a wider talent pool, and recruit candidates from various backgrounds who were not previously reached. Companies should also reassess their open opportunities because the way job descriptions are written and where they are posted plays an essential role in who applies. Equal opportunity statements at the end of job descriptions show candidates that a company values diversity and inclusion.
Georgetown University is not the only institution spearheading programs for systemic inequity awareness. Cheng praised numerous universities, such as UCLA, UCSF, and The Ohio State University, who have been at the forefront of implicit bias research and training. All three panelists are eager to continue the BRIC program. They hope that by scaling this bias awareness across Georgetown, there will always be individuals on committees who have been trained and can challenge their colleagues’ assumptions.









{kind=link}